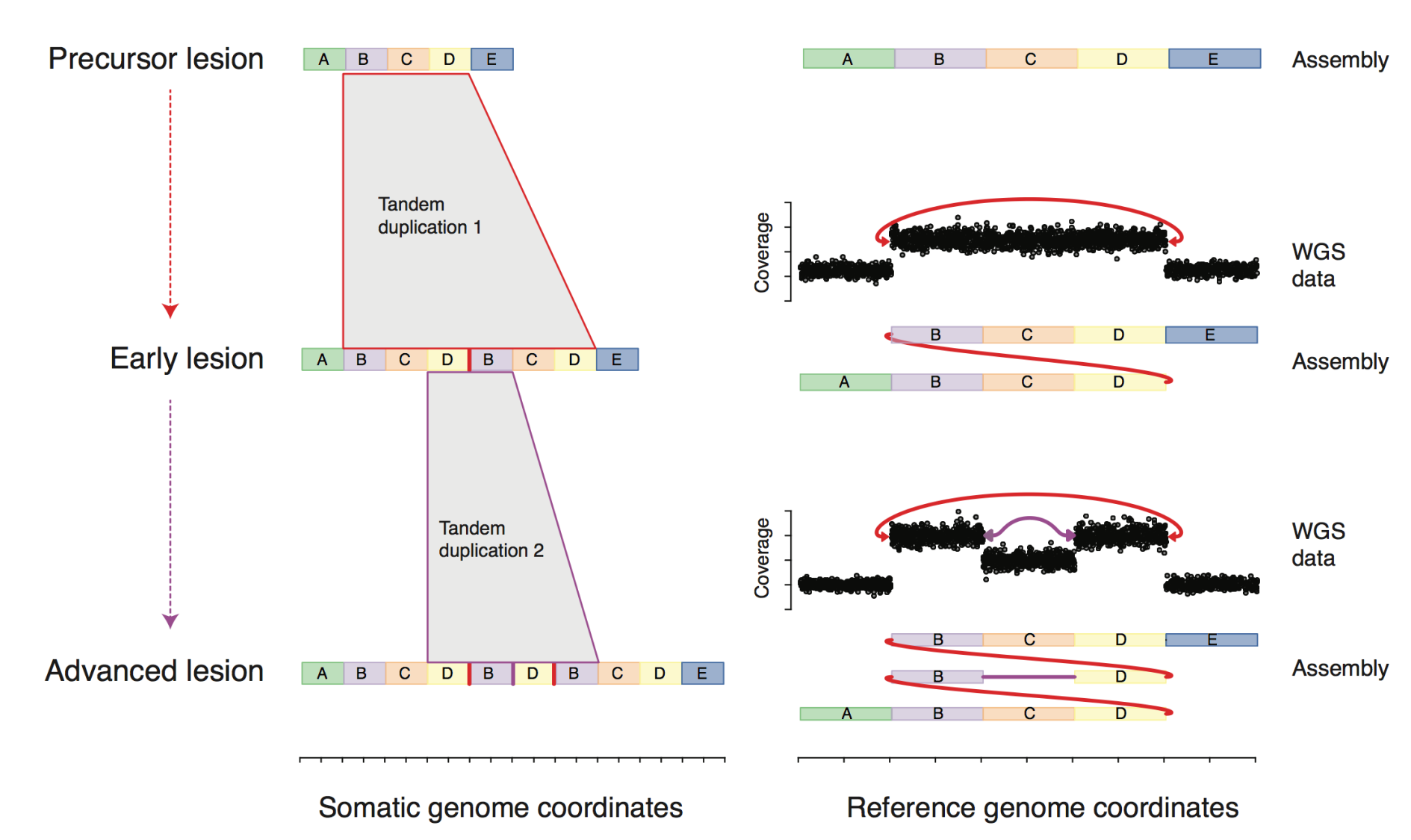
evolution of complex DNA rearrangements in cancer
A key challenge in analyzing cancer DNA rearrangements from short reads is defining what is an "event". However, with DNA rearrangements there is a fundamental ambiguity regarding the structure or history of a mutated region. In contrast, when we study a DNA substitution, we know both what the mutated locus looks like and how it arrived at its current state. Answering one question can help us answer the other, which is why we are using long-range sequencing technology, multi-biopsy sample sets, and computational modeling to characterize the structure and evolution of complex variants in cancer. This approach can be particularly useful in understanding large-scale rearrangement events, such as chromothripsis (chromosome shattering).
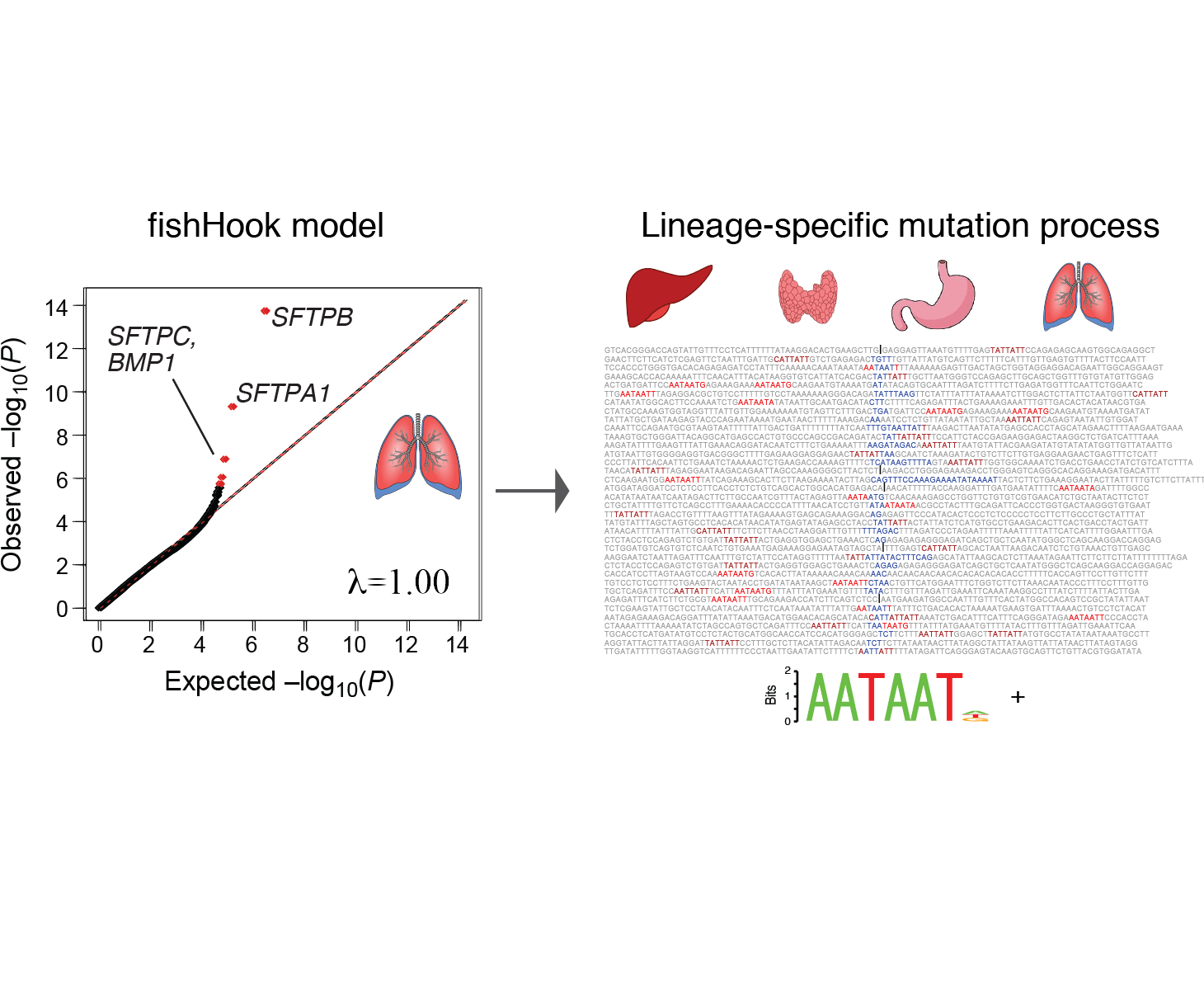
genomic footprints of cancer cell-of-origin
Work from our group and others have demonstrated footprints of the lineage history of cancer cells in their DNA, through correlation of mutation patterns with the transcriptomic and epigenetic features of the healthy tissue from which that cancer arose. Though these correlations preliminary have been drawn from transcriptomic and chromatin profiles of bulk healthy tissue, we are working to extend these correlations to other variant types and cell type specific epigenetic profiles. These patterns may provide an orthogonal piece of evidence to link cancer types to their presumed cell-of-origin.
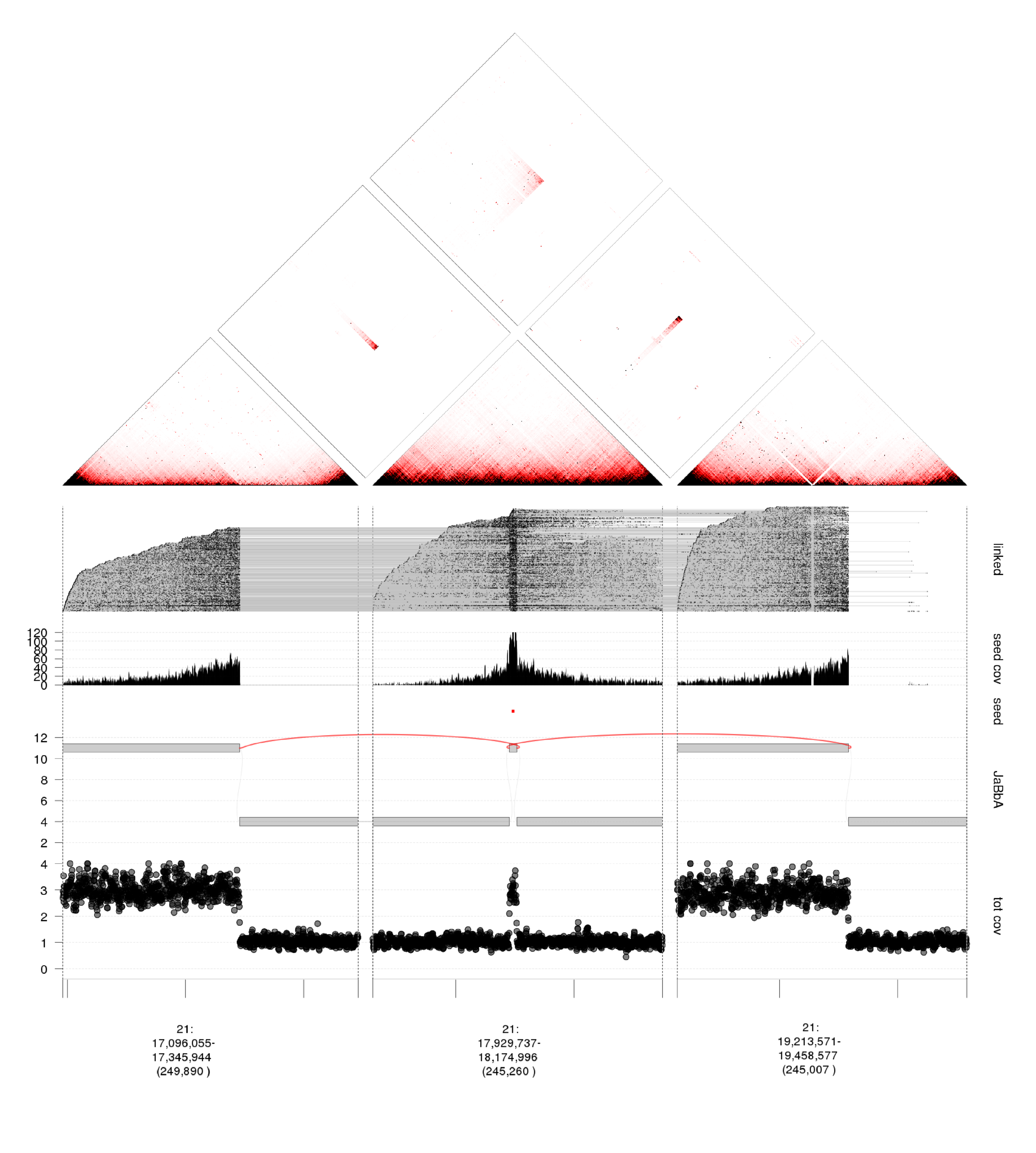
long range sequencing and cancer genome assembly
Short read (<300bp) sequences leave considerable ambiguity about the long-range structure or "phase" of altered loci. Phase is often essential for diagnosing the functional outcome of a given variant, and for inferring how that variant evolved. Such information is especially important in cancer genomes affected by complex clustered variants such as chromothripsis (chromosome shattering), kataegis (mutational showers), and chromoplexy (chains of balanced rearrangements). Using novel technologies, such as linked-read sequencing (10X genomics), we can computationally reconstruct the long range structure of variant loci in cancer, and infer novel event types. Analysis of these data requires development of novel algorithms, data formats, and visualization approaches, which the lab is actively engaged in.
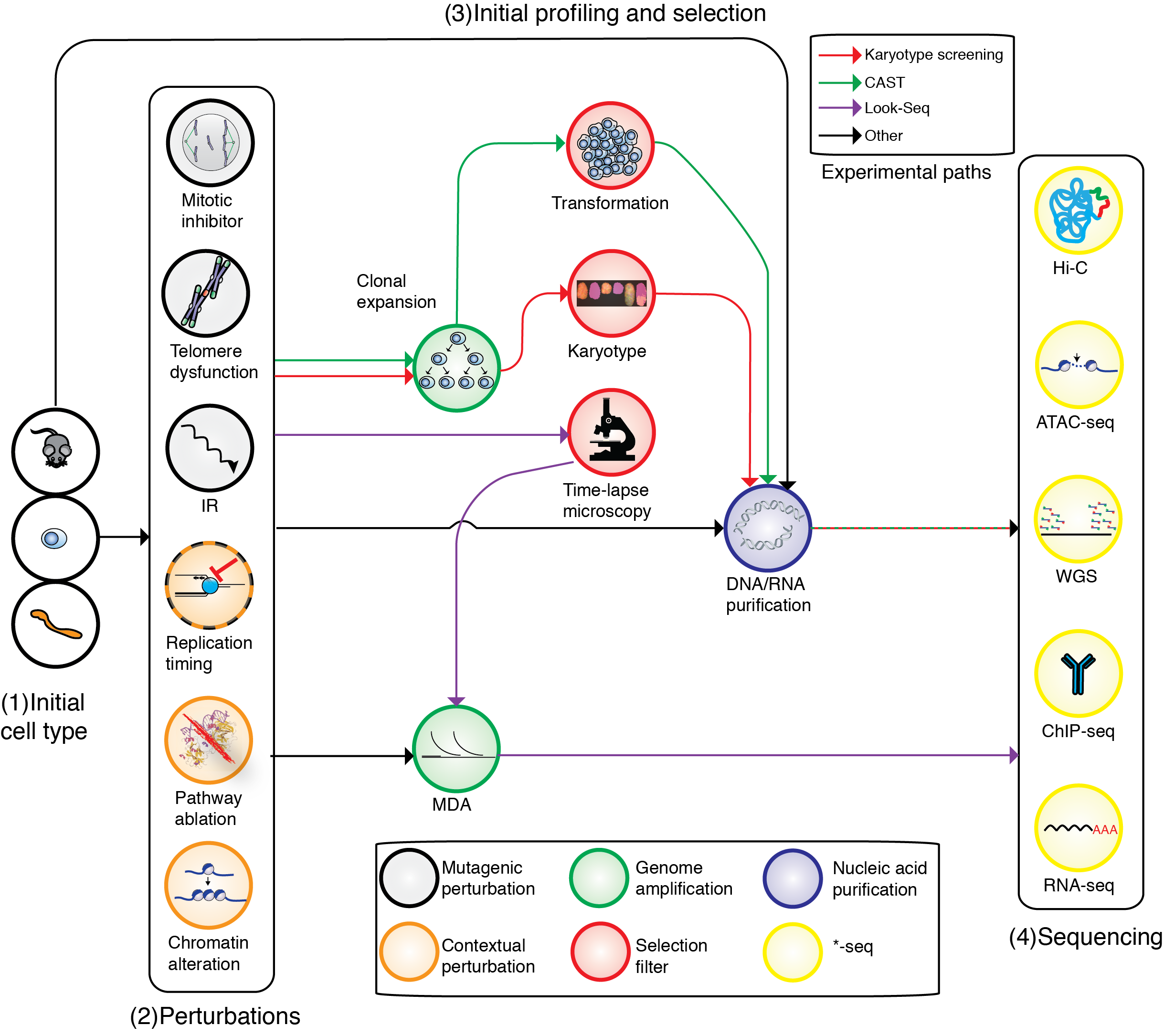
DNA signatures of somatic mutation processes and genome integrity defects
Every cancer genome provides a readout of mutational processes or DNA repair defects that are active in tumor cells or their healthy predecessors. Such phenomena have been extensively characterized for somatic SNV, but are less well known for indels and DNA rearrangements. The lab is actively pursuing novel analytic approaches in human cancer and engineered systems to study these processes and to infer their genetic or chemical determinants. Better understanding of these processes and repair defects can inform treatment choice for targeted therapy as well as classic chemotherapy and immunotherapy.

epigenetic consequences of somatic DNA variants
Somatic DNA variants that land outside of coding regions can alter how genes are transcribed and chromatin is organized in the nucleus. Unlike SNVs and indels, DNA rearrangements likely induce severe perturbations to the regulatory landscape of cells, though the nature of this impact is only beginning to be understood. We seek to characterize the impact of DNA rearrangements on the tumor epigenome through long-range sequencing and chromatin profiling of rearranged tumors. We hope this analysis will yield novel noncoding driver candidates in tumor subtypes where no known coding driver alteration has been discovered.